
Therapeutic Targeting of NLRP3 Inflammasome and JNK Pathways in Inflammation-Induced Insulin Resistance
-
Igbayilola Yusuff Dimeji


Department of Human Physiology, College of Medicine and Health Sciences, Baze University, Abuja, Nigeria
Hamidu Lawan JabbaDepartment of Human Physiology, College of Medicine and Health Sciences, Baze University, Abuja, Nigeria
Ngabea MurtalaDepartment of Medicine, Maitama District Hospital/College of Medicine, Baze University, Abuja, Nigeria
Adekola Saheed AyodejiDepartment of Chemical Pathology, Medical Laboratory Science Program, Faculty of Nursing and Allied Health Sciences, University of Abuja, Abuja, Nigeria
| Received 08 Jul, 2025 |
Accepted 01 Sep, 2025 |
Published 30 Sep, 2025 |
Inflammation is a physiological response of the organism to noxious stimuli, physical, chemical, or biological, with a chronic, low-grade inflammatory state, which suggests that inflammation may be an underlying mechanism by which obesity leads to insulin resistance. Among the array of molecular mechanisms implicated in this pathophysiological state, the NLRP3 inflammasome and the c-Jun N-terminal kinase (JNK) pathway have emerged as central mediators of immune–metabolic crosstalk. NLRP3 inflammasome and the c-Jun N-terminal kinase (JNK) signaling pathway are principal regulators of immunometabolism, the interface between immune cells and metabolism. The NLRP3 inflammasome, a protein complex, detects danger signals and induces the activation of caspase-1, leading to the secretion of inflammatory cytokines and pyroptosis. The JNK, a stress-activated kinase, participates in inflammation and metabolic derangements, such as insulin resistance. This review provides an extensive overview of the molecular mechanisms through which such pathways cause insulin resistance and summarizes recent pharmacologic strategies to control their activity. Highlighted are recent developments in addressing these inflammatory nodes, an assessment of preclinical and clinical data, and a discussion of the difficulties and potential paths for turning these discoveries into treatments.
INTRODUCTION
Several related metabolic diseases of metabolic, environmental, and/or genetic origin are commonly combined to form metabolic syndrome (MetS), insulin resistance syndrome (IRS), or syndrome X1. It comprises atherogenic dyslipidaemia (hypertriglyceridemia and/or low HDL cholesterol), insulin resistance, dysglycemia (reduced glucose tolerance and/or reduced fasting glucose), abdominal obesity2 and hypertension (at least three of them)3.
Over the past few decades, there has been a robust association between metabolic disease and inflammation, and consequently, the metaflammation hypothesis that chronic, low-grade, systemic inflammation caused by overnutrition and overloading with energy, has been suggested3-5. Cumulative evidence supports the notion that diabetes is an inflammatory disease at its essence. Type 1 diabetes mellitus (T1DM), traditionally referred to as an autoimmune disease consisting of immune-mediated β-cell destruction and relative insulin deficiency, has long been associated with inflammation6.
However, it was only in the early 1990s that type 2 diabetes mellitus (T2DM) also became implicated with inflammatory processes7. The T2DM is characterized by insulin resistance and impaired insulin secretion, along with chronic low-grade inflammation in peripheral organs such as adipose tissue, liver, and muscle8. Increasing evidence continues to testify to the relationship between obesity, insulin resistance, and inflammation8,9. As inflammation is playing a pivotal role in the disease pathogenesis, T2DM is also nowadays considered an immune-mediated disorder10. Furthermore, inflammation has also been found to be involved in a wide range of other metabolic diseases11. The current special issue brings together a set of original research papers and reviews on the regulatory role of inflammation in metabolic dysfunction.
The immune system’s defence against bacterial, viral, and fungal diseases depends heavily on the NLRP3 inflammasome12,13. On the other hand, when dysregulated, it has been linked to the aetiology of several inflammatory disorders, such as atherosclerosis, diabetes, gout, autoinflammatory diseases, Alzheimer’s disease, and cryopyrin-associated periodic syndromes (CAPS)14,15. Three domains make up the structure of NLRP3: The C-terminal leucine-rich repeat (LRR) domain, the central nucleotide-binding and oligomerization domain (NOD or NACHT), and the N-terminal pyrin domain (PYD)16,17. To start the inflammasome complex's assembly, the PYD domain engages with the ASC's pyrin domain18.
The ATPase activity has been recently identified as the target for MCC950, a widely employed selective NLRP3 inhibitor19. Contrary to expectations, recent evidence suggests that the LRR domain is unnecessary for NLRP3 autoinhibition as well as activation, in conflict with past suppositions about its role20.
The c-Jun N-terminal kinases (JNKs), a subfamily of the mitogen-activated protein kinases (MAPKs), play a role in cell survival and apoptosis due to extracellular and intracellular stressors21. The JNKs, or stress-activated protein kinases (SAPKs), are activated during exposure to bacterial toxins, environmental stress, and proinflammatory cytokines22. Consequently, JNK signaling contributes to the pathophysiology of numerous diseases through modulation of inflammatory responses, cell differentiation, growth, apoptosis, and survival23. The JNK family has three isoforms, namely JNK1, JNK2, and JNK3, which are encoded by the MAPK8, MAPK9, and MAPK10 genes, respectively24. These isoforms result in at least ten alternatively spliced variants: four for JNK1 and JNK2, and two for JNK325. The JNK1 and JNK2 are expressed ubiquitously, while JNK3 expression is limited to brain, heart, and testis26. Despite 83% sequence identity and redundant function, JNK2 prefers the substrate c-Jun over JNK1, implying differential regulatory functions27. The JNK1 and JNK2 are implicated in obesity, diabetes,28, immune dysfunction29, cancer30, and respiratory diseases31, whereas JNK3, primarily in the brain, is an attractive target for therapy against neurodegenerative diseases32,33. Novel research also names JNKs as important mediators of viral, bacterial, fungal, and parasitic infectious disorders34.
Two proteins that are key signaling modules, the NLRP3 inflammasome and JNK (c-Jun N-terminal kinase) pathway, play key roles in the bridges between metabolic disturbances such as obesity, insulin resistance, and type 2 diabetes and activation of innate immune responses. The NLRP3 inflammasome, a protein structure, senses danger signals like excess nutrients, reactive oxygen species (ROS), and mitochondrial injury, leading to activation of caspase-1 and release of proinflammatory cytokines IL-1β and IL-18. The inflammatory cascade is the cause of chronic low-grade inflammation that is the basis for most metabolic diseases. While this happens, the JNK pathway is also stimulated by the same stress stimuli, including free fatty acids and oxidative stress, and regulates the phosphorylation of insulin receptor substrates to disrupt insulin signaling and cause insulin resistance. In addition, JNK also elevates inflammatory gene expression
by activating transcription factors like AP-1 and c-Jun. The intersection of these pathways promotes metabolic inflammation and organ dysfunction of vital organs such as the liver, fat, and pancreas. Consequently, both NLRP3 inflammasome and JNK pathway have become promising pharmacological targets, with research focusing on small-molecule inhibitors, natural products, and biologics that can modulate their activity to decrease the advancement of metabolic disease and restore immune-metabolic homeostasis. This study investigated the molecular roles of the NLRP3 inflammasome and JNK signaling pathways in inflammation-induced insulin resistance and explored their potential as therapeutic targets for mitigating metabolic dysfunction.
NLRP3 inflammasome
Structure and activation: The NLRP3 protein is ubiquitously expressed in various cell types like myeloid cells, muscle cells, neurons, and endocrine cells35. In resting conditions, NLRP3 exists in an autoinhibited configuration that requires particular stimuli to turn on, thereby leading to the assembly of a large cytosolic inflammasome complex. In macrophages, NLRP3 activation is a tightly controlled two-step procedure involving priming and activation36. In the priming stage, cytokines like TNF-α and pattern recognition receptors like Toll-like receptors (TLRs) or NOD-like receptors activate the transcription factor NF-κB, which in turn raises the expression of important inflammasome components like pro-IL-1β, caspase-1, and NLRP3 itself37.
Then, NLRP3 undergoes many post-translational modifications such as ubiquitination, phosphorylation, and simulation maintaining the protein in a resting but signal-ready state38,39. During the second phase, after identification of a diverse array of cell stress stimuli, the NLRP3 inflammasome generates an active NLRP3, ASC (apoptosis-associated speck-like protein with a CARD), and procaspase-1 complex that allows proinflammatory cytokines IL-1β and IL-18 to process and be secreted40.
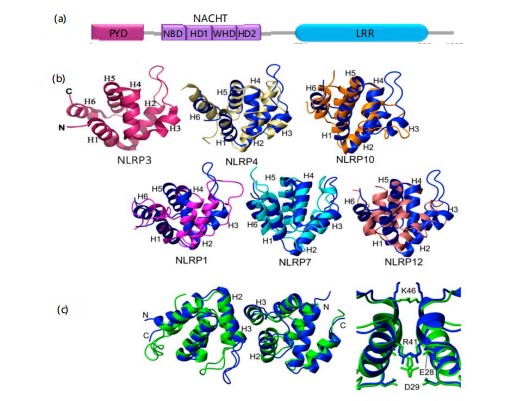
The NLRP3 contains three prominent domains (Fig. 1a), An N-terminal pyrin domain (PYD), a central NACHT domain, and a C-terminal leucine-rich repeat (LRR) domain41. Structural studies by X-ray crystallography and solution-state NMR have characterized the structure of the human NLRP3 PYD (NLRP3^PYD), which is a six-helix bundle (α1–α6) stabilized by five connecting loops an arrangement comparable to that of the PYD domains of NLRP1, NLRP4, NLRP7, NLRP10, and NLRP12 (Fig. 1b)42,43. Structural homology is strongest between NLRP3, NLRP4, and NLRP10, although there is minimal variation in helix length and direction42. The core structure of NLRP3^PYD is maintained by a central hydrophobic cluster spanning helices α1, α2, α4, α5, and α6, with the second hydrophobic surface stabilizing helix α3. Electrostatic surface analysis suggests potential interfaces for interaction with ASC^PYD or other Death Domain superfamily proteins42,43. A hydrophobic surface preserved by residues like I39, P40–P42, L57, and F61 is likely to mediate inflammasome assembly and caspase-1 activation. An unusual disulfide bond between C8 and C108 has been hypothesized to link ROS signaling with NLRP3 activation by destabilizing its autoinhibitory conformation42. Furthermore, the NMR and crystal structures of NLRP3^PYD exhibit high structural resemblance (RMSD 1.66 Å), with the central helices being rigid, indicated by low B-factors, whereas the α2-α3 loop and C-terminus are flexible (Fig. 1c). Recent cryo-electron microscopy has also revealed an earring-shaped conformation of the full-length NLRP3, which consists of a 12-repeat LRR domain folding into shape and a compact NACHT module comprising subdomains such as NBD, HD1, WHD, and HD241.
Role in insulin resistance: “Reduced insulin sensitivity” (IR) is the term used to describe the biological activity of normal insulin levels on target tissues in healthy humans. Insulin primarily affects peripheral tissues, such as muscle, liver, and adipose tissue, where it inhibits hepatic gluconeogenesis and promotes the absorption and utilization of glucose and glycogen production45,46. Defective insulin signalling pathways, or a series of anomalies in the signals sent to cells when insulin binds to the insulin receptor (INSR), are the primary cause of insulin resistance (IR)47.
|
Insulin resistance (IR) and low-grade chronic inflammation are the hallmark characteristics of type 2 diabetes mellitus (T2DM). Current evidence suggests that NLRP3 inflammasome is central to T2DM and IR pathogenesis48. Several glycolipid metabolites and their derivatives are capable of triggering the NLRP3 inflammasome. While dysregulation of glucose metabolism, i.e., hyperglycemia, has long been considered the primary stimulus for chronic inflammation, recent data suggest a close association between chronic inflammation and dysregulated glucose-stimulated NLRP3 inflammasome activation49. Glucose can trigger inflammasome activation through ATP/P2X purinergic receptor 4 pathways and by promoting expression of thioredoxin-interacting protein (TXNIP), a key cofactor in NLRP3 activation50,51. In hyperglycemic mouse models, pancreatic islet cells exposed to high extracellular glucose levels show increased NLRP3 activation and IL-1β release that induce IR52,53 Fig. 2). Additionally, saturated fatty acids such as palmitate induced the NLRP3 inflammasome, which results in the secretion of IL-1β and IL-18, and disrupts insulin signaling in gene knockout mice lacking NLRP3, Caspase-1, or ASC54. Stienstra et al.50 further demonstrated that blocking Caspase-1 improves insulin sensitivity in obese mice, which suggests the regulatory role of the inflammasome in adipocyte function and IR.
|
The T2DM is also associated with hypersecretion of islet amyloid polypeptide (IAPP), leading to amyloid deposits within pancreatic islets. Ingestion of such deposits by macrophages into lysosomes triggers NLRP3 activation and a series of inflammatory processes55,56. Fructose is also involved in the cause of IR of gestational diabetes via the NF-κB-NLRP3 pathway57. The NLRP3 inflammasome is involved mainly in IR through downstream signaling by IL-1β. Experiments have shown that deficiency of IL-1β protects against IR caused by a high-fat diet58. The NLRP3-deficient mice also show improved pancreatic β-cell function due to reduced IL-1β secretion. Glucose, free fatty acids, and ROS-induced endoplasmic reticulum stress activate the JNK pathway in obese individuals, while obesity-induced inflammation increases JNK and IKKβ activation further contributors to IR59. In models of obesity and T2DM, activation of NLRP3 inflammasome in adipose tissue in adipocytes or macrophages leads to increased IL-1β production and exacerbates IR. Inflammasome activation is inhibited specifically, diminishing β-cell inflammation and enhancing insulin sensitivity, potentially through mechanisms such as stimulation of the AMPK/NLRP3/HMGB1 signaling pathway60.
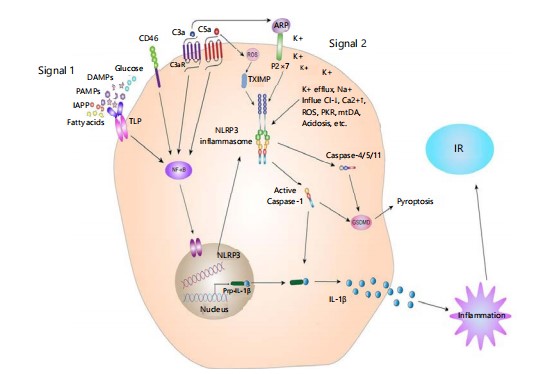
Schematic representation of two-step activation of the NLRP3 inflammasome. Signal 1 (priming) is the detection of pathogen- or damage-associated molecular patterns (PAMPs/DAMPs) by pattern recognition receptors such as TLRs, leading to NF-κB-dependent transcription of inflammasome subunits. Signal 2 (activation) includes stimuli such as ATP, ion fluxes, and ROS, followed by the assembly of NLRP3 complexes, caspase-1 activation, IL-1β maturation, pyro ptosis, and downstream inflammation precipitating insulin resistance (IR).
Genetic and preclinical evidence: The NLRP3 inflammasome activation has been implicated as a key mediator of chronic inflammation, a feature of obesity and insulin resistance (IR). Nutrient excess in obesity leads to the accumulation of danger-associated molecular patterns (DAMPs), which activate the NLRP3 inflammasome and thereby caspase-1 activation62,63. This cascade promotes the maturation and secretion of proinflammatory cytokines IL-1β and IL-18 by invading immune cells in obese adipose tissue. While numerous studies support a link between the activation of NLRP3 inflammasome and obesity or IR, some studies have been ambiguous64. Genetic evidence also supports this link because the NLRP3 intronic
variant rs10754555 was linked to elevated systemic inflammation, enhanced inflammasome activity, severe coronary artery disease, and elevated risk of mortality65. These findings illustrate the therapeutic promise of the NLRP3 inflammasome in cardiometabolic disease. In addition, a signaling pathway of YY1-mediated NLRP3 induction and PKCε activation has been revealed in recent research, suggesting the YY1-NLRP3-PKCε axis as a novel target for metabolic diseases such as non-alcoholic fatty liver disease (NAFLD)66.
In addition, more and more evidence suggests that mice lacking Nlrp3 or knocked down in Nlrp3 are resistant to diet-induced obesity, typically associated with insulin resistance (IR) and hepatic steatosis67. In addition to its canonical role in inflammasome activation, NLRP3 has also been reported to translocate into the nucleus, where it may have the potential to act as a transcriptional co-regulator with IRF4, binding directly to target genes involved in TH2 cell differentiation and in LPS/ATP-stimulated epithelial responses68,69. Myeloid-restricted NLRP3 activation within liver tissue has been linked with hepatocyte pyroptosis, liver inflammation, and fibrosis, all of which contribute to obesity and IR development70,71.
JNK pathway
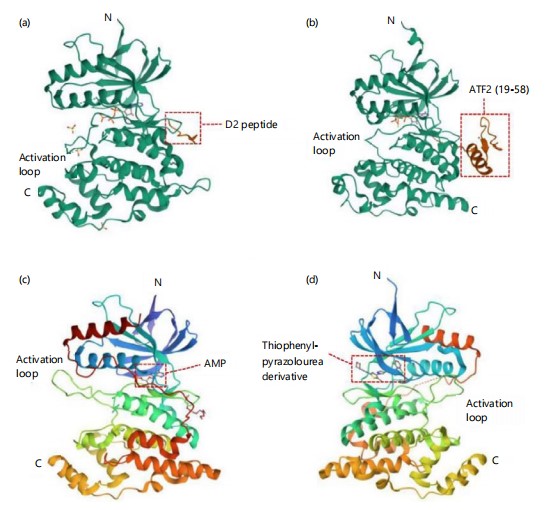
JNK structure and activation: The three isoforms of c-Jun N-terminal kinases (JNK1, JNK2, and JNK3) exhibit molecular weights of approximately 46 or 55 kDa due to differences such as a COOH-terminal extension in some variants72. Crystallographic analysis has revealed that all three isoforms share more than 90% sequence homology73,74. Structurally, JNK proteins consist of three main components: the N-terminal lobe, the C-terminal lobe, and a flexible linker segment. The N-lobe comprises multiple β-strands, while the C-lobe is rich in α-helices75,76. Functionally, the N-lobe contains glutamate-aspartate (ED) motifs, and the C-lobe includes the common docking (CD) domain75,76. The flexible linker region houses the kinase’s catalytic site, containing a Thr-Pro-Tyr motif that undergoes phosphorylation by MAP2K77.Additionally, JNK includes a conserved ATP-binding pocket and a kinase activation site, contributing to the challenge of developing selective small-molecule inhibitors75. Figure 3a-d displays the representative structure of human JNK1. More recently, the resolved structures of JNK1 bound to the MKK7 docking motif78, JNK1 in complex with ATF279, AMP-bound JNK280, and JNK3 in complex with thiophene–pyrazolourea inhibitors81. These findings enhance our understanding of JNK’s structural biology and will aid in the design of novel, isoform-specific JNK inhibitors.
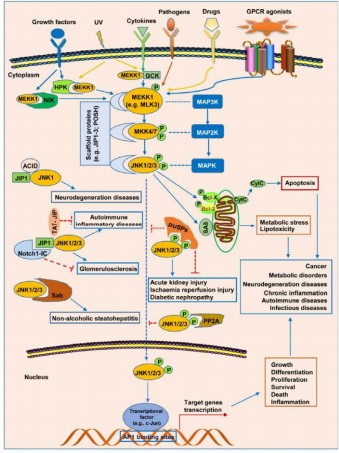
The c-Jun N-terminal kinase (JNK) is a member of the proline-directed serine/threonine kinases family and is stimulated by a wide range of extracellular stimuli (Fig. 4), such as cytokines, growth factors, reactive oxygen species (ROS), heat shock, shear stress, pathogens, and pharmacological agents82,83. As with the other mitogen-activated protein kinase (MAPK) pathways, the JNK signaling pathway also has three fundamental components: The MAP kinase kinase kinases (MAP3Ks), which include ASK1 or MEKK1; MAP kinase kinases (MAP2Ks), which are MKK4 and MKK7; and the MAPKs themselves, which include JNK1, JNK2, and JNK384. In response to stress signals, the MAP3Ks become activated, and in turn, they phosphorylate MKK4 and MKK7. These MAP2Ks phosphorylate and activate the JNK isoforms85. Activation of JNK, like other MAPKs, is through dual phosphorylation of a Thr-Pro-Tyr (T-X-Y) motif in a flexible peptide loop. Specifically, JNK1 activation is through phosphorylation of threonine-183 and tyrosine-18585. Although the general mechanism for activation of JNK is understood, it remains to be determined in most cases whether the increased JNK activity results from heightened kinase activity or increased levels of expression of the JNK proteins.
These stimulate STE20 protein homologs (HPK, NIK, GCK), which then phosphorylate MEKK1 or activate MAP3Ks (ASK1, TAK1, MLK3). These then activate MKK4 and MKK7, which then phosphorylate and activate JNK isoforms. JNK, on its part, phosphorylates non-nuclear proteins like c-Jun and ATF2, which induce gene transcription, as well as non-nuclear proteins, which modulate cell responses like growth, apoptosis, and inflammation. Protein complexes like JIP1-JNK1/2/3 are involved in JNK regulation and are linked with conditions like neurodegeneration and glomerulosclerosis.
|
Role in insulin resistance: Chronic low-grade inflammation triggered by obesity-induced FFA load activates JNK to lead to the phosphorylation of insulin receptor substrates (IRS) 1 and 2 on Ser/Thr residues86, and the dephosphorylation of IRS-1 on Tyr residues.87. This inhibits IRS from the activation of downstream PI3K and its interaction with the insulin receptor (IR). Thus, the PI3K-AKT insulin signaling pathway is blocked, reducing the sensitivity of target cells to insulin, which induces the body to release more insulin in an attempt to eliminate excess glucose88,89. In pancreatic β-cells, however, compensatory insulin secretion leads to hyperinsulinemia, increasing β-cell secretion stress90. It ultimately leads to hyperproliferation and apoptosis, culminating in insulin resistance and type 2 diabetes91. This pathway links JNK activity with obesity-related insulin resistance.
Several research have supported this, including Yang et al. who employed gene knockout technology to eliminate the JNK1 gene in mouse adipose tissue92. According to their findings, removing JNK1 inhibited the liver’s induced insulin resistance brought on by a high-fat diet. Insulin sensitivity was likewise enhanced by JNK2 suppression92. Using a macrophage-selective JNK-deficient mouse model93.
|
In obese inflammation, the function of adipose tissue macrophages increases the demand for glucose, which stimulates gluconeogenesis and fat breakdown, resulting in lipolysis and metabolic disturbances of glucose and lipids94. Inflammation also interrupts endothelial function and induces endothelial insulin resistance. Increased production of reactive oxygen species (ROS) also suppresses uncoupling protein (UCP) S transcription, lowering free radical scavenging and provoking oxidative stress95. Elevated ROS activate NF-κB and JNK signaling, which regulate nuclear and extranuclear substrates including AP-1 and NF-κB, impairing insulin action and GLUT4 translocation and resulting in insulin resistance.
Moreover, FFAs increase in the blood, getting metabolized to acyl-CoA and elevating diacylglycerol (DAG) levels. This activates PKCθ, altering IRS-1 phosphorylation and preventing PI3K activation and GLUT4 translocation to the cell surface, reducing insulin-stimulated glucose utilization. Other studies have shown that FFAs inhibit glucose uptake after stimulation by insulin, reducing glucose oxidation and glycolysis, hence reducing tissue sensitivity to insulin. The HFDs over-activate JNK, which causes insulin resistance.
|
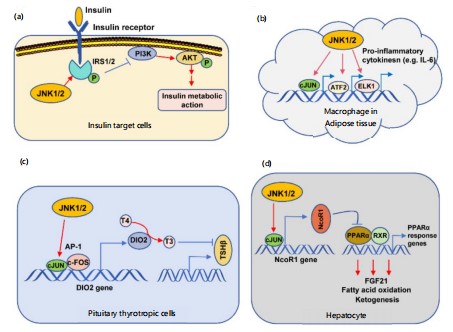
JNK’s role is underlined by a series of findings (Fig. 5): (1) hyper-activation of JNK in obesity, and JNK1-null mice are insulin-sensitive, (2) JNK1 knockout protects against obesity; (3) JNK2-null mice are insulin-sensitive; and (4) mutations in the JIP1 gene, the scaffold encoded for by JNK, are linked to type II diabetes96-99.
JNK contributes to type 2 diabetes through four methods, according to Solinas and Karin (2010): (1) phosphorylation of IRS1/2; (2) involvement in metabolic inflammation; (3) inhibition of the TSH-thyroid hormone axis; and (4) suppression of PPARa-FGF21 signalling. The improvement in insulin sensitivity with anti-TNFα medication provides clear evidence for the first mechanism Gluconeogenesis and insulin resistance are caused by TNFα-activated JNK's direct phosphorylation of IRS1/2, which interferes with PI3K-AKT signalling and insulin receptor interaction. The JNK phosphorylates IRS2,and JNK1 and JNK2 can phosphorylate IRS100-103.
Experimental evidence: Despite proof of inflammation's significant role in metabolic diseases, currently available anti-inflammatory therapies have not shown significant results although Wang et al.104 reported the role of L-arginine in immune modulation104. This validates the need to research novel targets for inflammation. Even though the direct inhibition of JNK may not be an effective treatment for human type 2 diabetes (T2D), a more complex disorder than the disease in animal models, preclinical studies show that JNK could be an effective drug target, particularly when combined with combination treatments for obesity and T2D. Genetic knockout models of JNK1 have shown beneficial effects against obesity-related insulin resistance, offering new evidence for the involvement of JNK in metabolically active tissues105. See Table 1.
| Table 1: | Molecular targets and therapeutic modulation of NLRP3 inflammasome and JNK pathways in inflammation-driven insulin resistance | |||
| Molecular pathway |
Key components |
Role in Inflammation -driven insulin resistance |
Potential molecular targets |
Therapeutic modulation strategies |
Therapeutic agents |
| NLRP3 inflammasome |
NLRP3 (NOD-like receptor family, pyrin domain containing 3), ASC, Caspase-1, IL-1β, IL-18 |
NLRP3 inflammasome activation is linked to the production of pro- inflammatory cytokines such as IL-1β and IL-18. Inhibition of NLRP3 reduces systemic inflammation and improves insulin sensitivity |
NLRP3 receptor -ASC (apoptosis- associated speck-like protein) Caspase-1-IL-1β |
NLRP3 inflammasome inhibitors to block cytokine release- Targeting ASC or Caspase-1 to inhibit inflammasome activation |
MCC950 (NLRP3 inhibitor)-CY -09 (NLRP3 inhibitor)-VX- 765 (Caspase- 1 inhibitor) -Anakinra (IL-1β antagonist) |
| JNK pathway | JNK (c-Jun N- terminal kinase), c-Jun, AP-1, TNF-α, IL-6 |
JNK activation contributes to the inflammatory response, causing increased insulin resistance. JNK-mediated phosphorylation of IRS-1 (insulin receptor substrate-1) reduces insulin signaling |
JNK (MAPK family)- c-Jun (transcription factor)-TNF-α (tumor necrosis factor-alpha) -IL-6 (interleukin 6) |
JNK inhibitors to reduce phosphorylation of IRS-1. Blocking TNF-α and IL-6 to attenuate the inflammatory response. Modulating c-Jun/AP-1 transcriptional activity |
SP600125 (JNK inhibitor)-Tofacitinib (JAK inhibitor, reduces IL-6 production- Etanercept (TNF-α inhibitor) |
| Inflammasome and JNK cross-talk |
Synergistic activation between NLRP3 and JNK in chronic inflammation. Both pathways enhance cytokine production and stress response |
Overlapping roles in increasing systemic inflammation, contributing to tissue dysfunction and insulin resistance |
NLRP3 and JNK inhibitors combined for synergistic anti- inflammatory effects |
Dual targeting strategies for inflammasome and JNK to reduce cytokine levels and improve insulin sensitivity |
Combination of MCC950 and SP600125 for effective inhibition. JNK inhibition combined with anti-IL-6 strategies |
| Molecular interactions |
NLRP3 inflammasome activation leads to the secretion of IL-1β. JNK signaling activates pro- inflammatory cytokines like TNF-α and IL-6 |
Both pathways contribute to systemic insulin resistance through chronic inflammation |
-Blocking crosstalk between NLRP3 and JNK using molecular inhibitors or gene silencing techniques |
Use of genetic approaches (siRNA, CRISPR) to block NLRP3 and JNK signaling. -Small molecule inhibitors or biologics for targeted therapy |
Small molecules targeting both pathways. Nanoparticle- delivered siRNA targeting both NLRP3 and JNK for more efficient therapy |
| Role in insulin sensitivity |
NLRP3-induced inflammation reduces insulin sensitivity through IL-1β release |
Insulin resistance is worsened by persistent activation of the inflammasome |
Targeting NLRP3 to reduce IL-1β- induced insulin resistance |
Blocking the inflammasome reduces systemic inflammation and improves insulin action |
Metformin (indirect modulation)- Glucagon-like peptide-1 (GLP-1) agonists (indirect modulation of inflammation) |
| NLRP3 inflammasome |
NLRP3 (NOD-like receptor family, pyrin domain containing 3), ASC, Caspase-1, IL-1β, IL-18 |
NLRP3 inflammasome activation is linked to the production of pro- inflammatory cytokines such as IL-1β and IL-18. Inhibition of NLRP3 reduces systemic inflammation and improves insulin sensitivity |
NLRP3 receptor -ASC (apoptosis- associated speck- like protein)- Caspase-1 - IL-1β |
NLRP3 inflammasome inhibitors to block cytokine release. Targeting ASC- or Caspase-1 to inhibit inflammasome activation |
MCC950 (NLRP3 inhibitor)-CY-09 (NLRP3 inhibitor)- VX-765 (Caspase-1 inhibitor)-Anakinra (IL-1β antagonist) |
| JNK pathway | JNK (c-Jun N-terminal kinase), c-Jun, AP-1, TNF-α, IL-6 |
JNK activation contributes to the inflammatory response, causing increased insulin resistance. JNK- mediated phosphorylation of IRS-1 (insulin receptor substrate-1) reduces insulin signaling |
JNK (MAPK family) -c-Jun (transcription factor)-TNF-α (tumor necrosis factor-alpha) -IL-6 (interleukin 6) |
JNK inhibitors to reduce phosphorylation of IRS-1. Blocking TNF-α and IL-6 to attenuate the inflammatory response. Modulating c-Jun/AP-1 transcriptional activity |
SP600125 (JNK inhibitor)-Tofacitinib (JAK inhibitor, reduces IL-6 production)- Etanercept (TNF-α inhibitor) |
| Inflammasome and JNK Cross-talk |
Synergistic activation between NLRP3 and JNK in chronic inflammation. Both pathways enhance cytokine production and stress response |
Overlapping roles in increasing systemic inflammation, contributing to tissue dysfunction and insulin resistance |
NLRP3 and JNK inhibitors combined for synergistic anti- inflammatory effects |
Dual targeting strategies for inflammasome and JNK to reduce cytokine levels and improve insulin sensitivity |
Combination of MCC950 and SP600125 for effective inhibition. JNK inhibition combined with anti-IL-6 strategies |
Pharmacological modulation of NLRP3 and JNK pathways
NLRP3 inhibitors: The MCC950 is a specific small-molecule inhibitor of the NLRP3 inflammasome, which is an essential part of the innate immune system that is implicated in the triggering of inflammatory responses. Through inhibition of the ATPase activity of NLRP3, MCC950 prevents the assembly and activation of the inflammasome complex that activates pro-inflammatory cytokines such as IL-1β and IL-1840. These cytokines play a central role in chronic inflammation, which is widely observed in obesity and metabolic diseases like insulin resistance and type 2 diabetes (T2D). Preclinical studies have shown that MCC950 can mitigate systemic inflammation in animal models, particularly in high-fat diet (HFD)-induced obese mice. The reduction in inflammation is also accompanied by significant improvement in glucose metabolism and a considerable enhancement in insulin resistance, signaling MCC950's therapeutic potential for metabolic diseases associated with chronic inflammation106.
Other than MCC950, other NLRP3 inflammasome inhibitors for their therapeutic potential in metabolic disease are being investigated. Two such agents that have shown promise in preclinical and early clinical trials are OLT1177 and dapansutrile. These agents selectively block the NLRP3 inflammasome, reducing inflammatory responses involved in insulin resistance and metabolic dysfunction. Inhibitors like MCC950 aim to attack the inflammatory pathways underlying diseases like obesity and T2D107. Research suggests that such inhibitors are in the process, but at the very beginning, it is already clear that NLRP3 inflammasome inhibition can open a new avenue in the treatment of metabolic disease, especially in patients with whom the anti-inflammatory component of the disease is not adequately addressed by conventional drugs.
JNK Inhibitors: The SP600125, which was one of the first identified JNK inhibitors, has been widely used in preclinical studies to examine the role of JNK in various diseases, including metabolic diseases75. The compound competes with ATP at the catalytic site of JNK, inhibiting its activation and downstream signaling. SP600125 has been found to be effective in improving insulin resistance in animal models, showing potential for the treatment of obesity-associated metabolic disorders108. By inhibiting JNK’s activity, it helps to restore insulin signaling pathways in, particular, insulin target tissues like the liver, fat tissue, and skeletal muscle. Despite its encouraging action in experimental models, SP600125's clinical application is thwarted by its off-target activities and less-than-ideal pharmacokinetic characteristics, such as poor bioavailability and rapid metabolism, which significantly undermine its therapeutic potential in humans.
Because of these limitations, more selective JNK inhibitors with better pharmacokinetic profiles and specificity have been developed. CC-930 (tanzisertib), for instance, is in clinical trials for fibrotic and inflammatory conditions like systemic sclerosis and idiopathic pulmonary fibrosis109. This selective JNK inhibitor has shown effectiveness in preclinical models by inhibiting certain JNK isoforms without the broad off-target effects of SP600125. With its selective action and better pharmacokinetics, CC-930 might also have the potential to treat metabolic diseases, such as type 2 diabetes and obesity, through the diminution of inflammation and enhancement of insulin sensitivity. Its continued development highlights the need to further optimize JNK inhibition approaches to better achieve both efficacy and safety for clinical application in the treatment of metabolic disease.
Dual and indirect modulators: Several pharmacological agents, including metformin and GLP-1 receptor agonists, have been discovered to inhibit both the NLRP3 inflammasome and JNK pathways indirectly, as a dual therapeutic approach in the treatment of inflammation and insulin resistance117. Metformin, used extensively in the treatment of type 2 diabetes, has been discovered to reduce systemic inflammation by downregulating the activation of these pathways, hence improving insulin sensitivity110. Similarly, GLP-1 receptor agonists such as liraglutide and semaglutide not only enhance glucose metabolism but also possess anti-inflammatory effects through the inhibition of NLRP3 and JNK activity. Other than these drug molecules, natural compounds such as curcumin, resveratrol, and berberine have been investigated for their activities in modulating these pathways. Curcumin, turmeric’s bioactive component, has been demonstrated to inhibit NLRP3 inflammasome activation and JNK-mediated inflammation, while resveratrol, a grape and red wine polyphenol, and berberine, a plant alkaloid, have both demonstrated encouraging anti-inflammatory and insulin-sensitizing effects through targeting these key signaling pathways111. These natural compounds are an attractive option for nutraceutical interventions against metabolic disease, providing an adjunct approach to traditional pharmacological treatments.
CHALLENGES AND FUTURE PERSPECTIVES
Although there is great promise in targeting the NLRP3 inflammasome and JNK signaling pathways to treat diseases like insulin resistance and chronic inflammation, there are many challenges that must be overcome before these can be implemented widely in clinical settings. Safety and selectivity are among the biggest concerns. The majority of current inhibitors of these pathways are non-selective and therefore may affect other signaling cascades and initiate off-target effects. For example, early JNK inhibitors such as SP600125, while capable of correcting insulin resistance in animal models, are inhibitors of other kinases too and produce unwanted side effects112. Similarly, NLRP3 inhibitors such as MCC950, while extremely promising in preclinical models, can also exert unexpected interactions with other components of the immune system, having safety issues when used for a long time. Achieving high specificity in inhibitor design is crucial in reducing the potential for side effects and ensuring that the therapies affect only the pathways that are accountable for disease advancement113-115.
Tissue specificity is a second important challenge. Both NLRP3 inflammasome and JNK signaling pathways play significant roles in immune surveillance, cell homeostasis, and other physiological processes116-118. Systemic inhibition of such pathways might interfere with their normal function in intact tissues and lead to unintended consequences. For instance, JNK is tasked with the regulation of cellular stress responses and apoptosis, both important for immunity and tissue repair119. Similarly, NLRP3 initiates immune response against infection, and inhibiting it would predispose the body to failure at clearing pathogens120. Accordingly, therapeutics that are disrupting these pathways may have wider implications for immune system function. Finally, despite strong preclinical efficacy in models of animal systems, clinical efficacy has yet to be proven. Human trials have been few and although promising at the preclinical level, follow-up in terms of clinical outcome has not been assured. This discrepancy highlights the need for larger clinical trials to further define the therapeutic potential and limitations of targeting these pathways in humans.
CONCLUSION
The NLRP3 inflammasome and JNK signaling pathway are core to insulin resistance induction, which is the core feature of metabolic syndrome. Low-grade chronic inflammation caused by stimuli such as excess adiposity, free fatty acids, and inflammatory cytokines activates both NLRP3 inflammasome and the JNK pathway, leading to IRS phosphorylation and disruption of insulin signaling pathways. This results in glucose metabolic dysfunction, further increasing insulin resistance. Inhibition of these pathways offers one mechanism to treat both inflammation and metabolic disease in conditions like type 2 diabetes and obesity. Preclinical models have identified several drugs that can inhibit the NLRP3 inflammasome and JNK pathway, increasing insulin sensitivity and inhibiting systemic inflammation. However, the application of these findings in clinical settings remains a challenge, as the majority of these inhibitors have exhibited off-target activities or poor selectivity in human trials. To advance, further research will be necessary to clarify further the complex interactions between immunoprotective mechanisms and metabolic regulation. Broad investigations into tissue-selective actions, extended pharmacologic safety over the long term, and the timing of treatment will be critical to therapy aimed at the NLRP3 inflammasome and JNK signaling pathway.
SIGNIFICANCE STATEMENT
This study addresses a critical gap in understanding how chronic inflammation drives insulin resistance, a key feature of metabolic disorders such as type 2 diabetes. While both NLRP3 inflammasome activation and JNK pathway signaling are known contributors, their combined effects have remained unclear. Here, the authors show that dual pharmacological inhibition of these pathways significantly enhances insulin sensitivity in both cell models and high-fat diet-induced diabetic mice. The approach reduces pro-inflammatory cytokine production (IL-1β, TNF-α), restores insulin signaling via IRS-1/Akt phosphorylation, and improves glucose tolerance beyond the effects of targeting either pathway alone. These findings reveal that NLRP3 and JNK redundantly mediate inflammation-induced insulin resistance, and that simultaneous targeting yields synergistic therapeutic effects. This insight opens avenues for developing combination therapies aimed at reversing early-stage insulin resistance, potentially preventing progression to type 2 diabetes. Moreover, this strategy may hold promise for treating related chronic inflammatory conditions like atherosclerosis and nonalcoholic fatty liver disease, which share overlapping inflammatory mechanisms.
REFERENCES
- Oda, E., 2018. Historical perspectives of the metabolic syndrome. Clin. Dermatol., 36: 3-8.
- Ehrampoush, E., R. Homayounfar, S.H. Davoodi, H. Zand, A. Askari and S.A. Kouhpayeh, 2016. Ability of dairy fat in inducing metabolic syndrome in rats. SpringerPlus, 5.
- Gisondi, P., A.C. Fostini, I. Fossà, G. Girolomoni and G. Targher, 2018. Psoriasis and the metabolic syndrome. Clin. Dermatol., 36: 21-28.
- Hotamisligil, G.S., 2006. Inflammation and metabolic disorders. Nature, 444: 860-867.
- Zhazykbayeva, S., S. Pabel, A. Mügge, S. Sossalla and N. Hamdani, 2020. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys. Rev., 12: 947-968.
- Durá-Travé, T. and F. Gallinas-Victoriano, 2025. Type 1 diabetes mellitus and vitamin D. Int. J. Mol. Sci., 26.
- Hotamisligil, G.S., N.S. Shargill and B.M. Spiegelman, 1993. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science, 259: 87-91.
- Winer, D.A., S. Winer, L. Shen, P.P. Wadia and J. Yantha et al., 2011. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med., 17: 610-617.
- Gregor, M.F. and G.S. Hotamisligil, 2011. Inflammatory mechanisms in obesity. Annu. Rev. Immunol., 29: 415-445.
- Tsai, S., X. Clemente-Casares, X.S. Revelo, S. Winer and D.A. Winer, 2015. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes, 64: 1886-1897.
- Chen, J., Y. Yang and W. Kong, 2022. Cross talk between inflammation and metabolic disorders. Mediators Inflammation, 2022.
- Kim, J.J. and E.K. Jo, 2013. NLRP3 inflammasome and host protection against bacterial infection. J. Korean Med. Sci., 28: 1415-1423.
- Menu, P. and J.E. Vince, 2011. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol., 166: 1-15.
- Bonnekoh, H., M. Butze, T. Kallinich, N. Kambe, G. Kokolakis and K. Krause, 2020. Spectrum of genetic autoinflammatory diseases presenting with cutaneous symptoms. Acta Dermato Venereologica, 100: 140-151.
- Ozaki, E., M. Campbell and S.L. Doyle, 2015. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflam. Res., 8: 15-27.
- Duan, Y., J. Wang, J. Cai, N. Kelley and Y. He, 2022. The leucine-rich repeat (LRR) domain of NLRP3 is required for NLRP3 inflammasome activation in macrophages. J. Biol. Chem., 298.
- Cescato, M., Y.Y.J. Zhu, L. le Corre, B.F. Py, S. Georgin-Lavialle and M.P. Rodero, 2024. Implication of the LRR domain in the regulation and activation of the NLRP3 inflammasome. Cells, 13.
- Bhat, E.A., N. Sajjad, J.A. Tantray, Y.Y. Hor and I.A. Rather, 2021. In vitro complex formation of human PYRIN domain-only protein 3 prevented by self-oligomerization of ASC PYD domain. Saudi J. Biol. Sci., 28: 1607-1614.
- Tapia-Abellán, A., D. Angosto-Bazarra, H. Martínez-Banaclocha, C. de Torre-Minguela, J.P. Cerón-Carrasco et al., 2019. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol., 15: 560-564.
- Hafner-Bratkovič, I., P. Sušjan, D. Lainšček, A. Tapia-Abellán and K. Cerović et al., 2018. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun., 9.
- Tao, W., W. Xu, X. Li, X. Zhang, C. Li and M. Guo, 2024. Characterization of c-Jun N-terminal kinase (JNK) gene reveals involvement of immune defense against Vibrio splendidus infection in Apostichopus japonicus. Fish Shellfish Immunol., 153.
- Shashikanth, N., O. Alaidi, L. Basa, S. Taank, R. Rao and J. Seetharaman, 2025. Role of c-Jun N-terminal kinases on a stressed epithelium: Time for testing isoform specificity. Biology, 14.
- Hammouda, M.B., A.E. Ford, Y. Liu and J.Y. Zhang, 2020. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells, 9.
- Priego, M., L. Noriega, S. Kalinin, L.M. Hoffman, D.L. Feinstein and G. Morfini, 2023. Genetic deletion of c-Jun amino-terminal kinase 3 (JNK3) modestly increases disease severity in a mouse model of multiple sclerosis. J. Neuroimmunol., 382.
- Figuera-Losada, M. and P.V. LoGrasso, 2012. Enzyme kinetics and interaction studies for human JNK1β1 and substrates activating transcription factor 2 (ATF2) and c-Jun N-terminal kinase (c-Jun). J. Biol. Chem., 287: 13291-13302.
- Conze, D., T. Krahl, N. Kennedy, L. Weiss and J. Lumsden et al., 2002. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J. Exp. Med., 195: 811-823.
- Tournier, C., 2013. The 2 faces of JNK signaling in cancer. Genes Cancer, 4: 397-400.
- Das, M., W.M. Zawada, J. West and K.R. Stenmark, 2018. JNK2 regulates vascular remodeling in pulmonary hypertension. Pulm. Circ. 8: 1-13.
- Musi, C.A., G. Agrò, F. Santarella, E. Iervasi and T. Borsello, 2020. JNK3 as therapeutic target and biomarker in neurodegenerative and neurodevelopmental brain diseases. Cells, 9.
- Wu, Y., Y. Zhao, Z. Guan, S. Esmaeili, Z. Xiao and D. Kuriakose, 2024. JNK3 inhibitors as promising pharmaceuticals with neuroprotective properties. Cell Adhes. Migr., 18: 1-11.
- Salazar, F., E. Bignell, G.D. Brown, P.C. Cook and A. Warris, 2021. Pathogenesis of respiratory viral and fungal coinfections. Clin. Microbiol. Rev., 35.
- Jha, S., W.J. Brickey and J.P.Y. Ting, 2017. Inflammasomes in myeloid cells: Warriors within. Microbiol. Spectrum, 5.
- Biasizzo, M. and N. Kopitar-Jerala, 2020. Interplay between NLRP3 inflammasome and autophagy. Front. Immunol., 11.
- Liu, T., L. Zhang, D. Joo and S.C. Sun, 2017. NF-κB signaling in inflammation. Signal Transduction Targeted Ther., 2.
- O’Keefe, M.E., G.R. Dubyak and D.W. Abbott, 2024. Post-translational control of NLRP3 inflammasome signaling. J. Biol. Chem., 300.
- Kim, Y., S. Lee and Y.H. Park, 2024. NLRP3 negative regulation mechanisms in the resting state and its implications for therapeutic development. Int. J. Mol. Sci., 25. https://doi.org/10.3390/ijms25169018
- Kelley, N., D. Jeltema, Y. Duan and Y. He, 2019. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci., 20. https://doi.org/10.3390/ijms20133328
- Sharif, H., L. Wang, W.L. Wang, V.G. Magupalli and L. Andreeva et al., 2019. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature, 570: 338-343.
- Bae, J.Y. and H.H. Park, 2011. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem., 286: 39528-39536.
- Oroz, J., S. Barrera-Vilarmau, C. Alfonso, G. Rivas and E. de Alba, 2016. ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces. J. Biol. Chem., 291: 19487-19501.
- Sharma, M. and E. de Alba, 2021. Structure, activation and regulation of NLRP3 and AIM2 inflammasomes. Int. J. Mol. Sci., 22.
- Igbayilola, Y.D. and M.G. Gujja, 2024. Alpha-amylase and alpha-glucosidase upregulated glucose homeostasis in high-fat fed Wistar rats supplemented with cocoa flavonoid-rich aqueous. Food Biosci., 59.
- Dimeji, I.Y., A.O. Samson, A. Kayode, A. Ikponwosa, W.O. Dada and O.F. Adesina, 2023. Glucometabolic response to walnut (Juglans regia L.) supplementation during gestation and/or lactation in offspring of Sprague-Dawley rats. Singapore J. Sci. Res., 13: 79-87.
- Szablewski, L., 2024. Changes in cells associated with insulin resistance. Int. J. Mol. Sci., 25.
- Ding, S., S. Xu, Y. Ma, G. Liu, H. Jang and J. Fang, 2019. Modulatory mechanisms of the NLRP3 inflammasomes in diabetes. Biomolecules, 9.
- Mohamed, I.N., L. Li, S. Ismael, T. Ishrat and A.B. El-Remessy, 2021. Thioredoxin interacting protein, a key molecular switch between oxidative stress and sterile inflammation in cellular response. World J. Diabetes, 12: 1979-1999.
- Qayyum, N., M. Haseeb, M.S. Kim and S. Choi, 2021. Role of thioredoxin-interacting protein in diseases and its therapeutic outlook. Int. J. Mol. Sci., 22.
- Mo, Y., L. Mo, Y. Zhang, Y. Zhang, J. Yuan and Q. Zhang, 2023. High glucose enhances the activation of NLRP3 inflammasome by ambient fine particulate matter in alveolar macrophages. Part. Fibre Toxicol., 20.
- Meyers, A.K. and X. Zhu, 2020. The NLRP3 inflammasome: Metabolic regulation and contribution to inflammaging. Cells, 9.
- Stienstra, R., L.A.B. Joosten, T. Koenen, B. van Tits and J.A. van Diepen et al., 2010. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab., 12: 593-605.
- Roham, P.H., S.N. Save and S. Sharma, 2022. Human islet amyloid polypeptide: A therapeutic target for the management of type 2 diabetes mellitus. J. Pharm. Anal., 12: 556-569.
- Liu, Y., Y. Wei, L. Wu, X. Lin and R. Sun et al., 2022. Fructose induces insulin resistance of gestational diabetes mellitus in mice via the NLRP3 inflammasome pathway. Front. Nutr., 9.
- McGillicuddy, F.C., C.M. Reynolds, O. Finucane, E. Coleman and K.A. Harford et al., 2013. Long-term exposure to a high-fat diet results in the development of glucose intolerance and insulin resistance in interleukin-1 receptor I-deficient mice. Am. J. Physiol. Endocrinol. Metab., 305: E834-E844.
- Iqra, H., S.R. Masoodi, S.A. Mir, M. Nabi, K. Ghazanfar and B.A. Ganai, 2015. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes, 6: 598-612.
- Litwiniuk, A., W. Bik, M. Kalisz and A. Baranowska-Bik, 2021. Inflammasome NLRP3 potentially links obesity-associated low-grade systemic inflammation and insulin resistance with alzheimer’s disease. Int. J. Mol. Sci., 22.
- Lu, S., Y. Li, Z. Qian, T. Zhao, Z. Feng, X. Weng and L. Yu, 2023. Role of the inflammasome in insulin resistance and type 2 diabetes mellitus. Front. Immunol., 14.
- Rheinheimer, J., B.M. de Souza, N.S. Cardoso, A.C. Bauer, D. Crispim, 2017. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism, 74: 1-9.
- Wani, K., H. AlHarthi, A. Alghamdi, S. Sabico and N.M. Al-Daghri, 2021. Role of NLRP3 inflammasome activation in obesity-mediated metabolic disorders. Int. J. Environ. Res. Public Health, 18.
- Jorquera, G., J. Russell, M. Monsalves-Álvarez, G. Cruz and D. Valladares-Ide et al., 2021. NLRP3 Inflammasome: Potential role in obesity related low-grade inflammation and insulin resistance in skeletal muscle. Int. J. Mol. Sci., 22.
- Schunk, S.J., M.E. Kleber, W. März, S. Pang and S. Zewinger et al., 2021. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur. Heart J., 42: 1742-1756.
- Qin, W. and J. Weng, 2023. Hepatocyte NLRP3 interacts with PKCε to drive hepatic insulin resistance and steatosis. Sci. Bull., 68: 1413-1429.
- Zhu, L. and L. Liu, 2022. New insights into the interplay among autophagy, the NLRP3 inflammasome and inflammation in adipose tissue. Front. Endocrinol., 13.
- Accogli, T., C. Hibos and F. Vegran, 2023. Canonical and non-canonical functions of NLRP3. J. Adv. Res., 53: 137-151.
- Zheng, J., L. Yao, Y. Zhou, X. Gu and C. Wang et al., 2021. A novel function of NLRP3 independent of inflammasome as a key transcription factor of IL-33 in epithelial cells of atopic dermatitis. Cell Death Dis., 12.
- Frissen, M., L. Liao, K.M. Schneider, S. Djudjaj and J. Haybaeck et al., 2021. Bidirectional role of NLRP3 during acute and chronic cholestatic liver injury. Hepatology, 73: 1836-1854.
- Mridha, A.R., A. Wree, A.A.B. Robertson, M.M. Yeh and C.D. Johnson et al., 2017. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol., 66: 1037-1046.
- Maik-Rachline, G., I. Wortzel and R. Seger, 2021. Alternative splicing of MAPKs in the regulation of signaling specificity. Cells, 10.
- Parker, J.A. and C. Mattos, 2018. The K-Ras, N-Ras, and H-Ras isoforms: Unique conformational preferences and implications for targeting oncogenic mutants. Cold Spring Harbor Perspect. Med., 8.
- Stachowski, T.R., S. Nithianantham, M. Vanarotti, K. Lopez and M. Fischer, 2023. Pan-HSP90 ligand binding reveals isoform-specific differences in plasticity and water networks. Protein Sci., 32.
- Yan, H., L. He, de Lv, J. Yang and Z. Yuan, 2024. The role of the dysregulated JNK signaling pathway in the pathogenesis of human diseases and its potential therapeutic strategies: A comprehensive review. Biomolecules, 14.
- Pei, J. and Q. Cong, 2023. Computational analysis of regulatory regions in human protein kinases. Protein Sci., 32.
- Sok, P., G. Gógl, G.S. Kumar, A. Alexa and N. Singh et al., 2020. MAP kinase-mediated activation of RSK1 and MK2 substrate kinases. Structure, 28: 1101-1113.E5.
- Kragelj, J., A. Palencia, M.H. Nanao, D. Maurin, G. Bouvignies, M. Blackledge and M.R. Jensen, 2015. Structure and dynamics of the MKK7–JNK signaling complex. Proc. Natl. Acad. Sci. U.S.A., 112: 3409-3414.
- Kirsch, K., A. Zeke, O. Tőke, P. Sok and A. Sethi et al., 2020. Co-regulation of the transcription controlling ATF2 phosphoswitch by JNK and p38. Nat. Commun., 11.
- Lu, W., Y. Liu, Y. Gao, Q. Geng and D. Gurbani et al., 2023. Development of a covalent inhibitor of c-jun N-terminal protein kinase (JNK) 2/3 with selectivity over JNK1. J. Med. Chem., 66: 3356-3371.
- Rajapaksa, N.S., 2021. In This Issue, Volume 12, Issue 1. ACS Med. Chem. Lett., 12: 1-2.
- Zheng, K., S. Iqbal, P. Hernandez, H. Park, P.V. LoGrasso and Y. Feng, 2014. Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives. J. Med. Chem., 57: 10013-10030.
- Craige, S.M., K. Chen, R.M. Blanton, J.F. Keaney and S. Kant, 2019. JNK and cardiometabolic dysfunction. Biosci. Rep., 39.
- Castro-Torres, R., O. Busquets, A. Parcerisas, E. Verdaguer and J. Olloquequi et al., 2020. Involvement of JNK1 in neuronal polarization during brain development. Cells, 9.
- Feng, J., S. Lu, B. Ou, Q. Liu and J. Dai et al., 2020. The role of JNk signaling pathway in obesity-driven insulin resistance. Diabetes Metab. Syndr. Obesity: Targets Ther., 13: 1399-1406.
- Báez, A.M., G. Ayala, A. Pedroza-Saavedra, H.M. González-Sánchez and L.C. Amparan, 2024. Phosphorylation codes in IRS-1 and IRS-2 are associated with the activation/inhibition of insulin canonical signaling pathways. Curr. Issues Mol. Biol., 46: 634-649.
- Zheng, M. and P. Wang, 2021. Role of insulin receptor substance-1 modulating PI3K/Akt insulin signaling pathway in Alzheimer’s disease. 3 Biotech, 11.
- Petersen, M.C. and G.I. Shulman, 2018. Mechanisms of insulin action and insulin resistance. Physiol. Rev., 98: 2133-2223.
- Dludla, P.V., S.E. Mabhida, K. Ziqubu, B.B. Nkambule and S.E. Mazibuko-Mbeje et al., 2023. Pancreatic β-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes, 14: 130-146.
- Galicia-Garcia, U., A. Benito-Vicente, S. Jebari, A. Larrea-Sebal and H. Siddiqi et al., 2020. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci., 21.
- Yang, H., C. Zhang, W. Kim, M. Shi and M. Kiliclioglu et al., 2024. Multi-tissue network analysis reveals the effect of JNK inhibition on dietary sucrose-induced metabolic dysfunction in rats. Comput. Syst. Biol. Med., 13.
- Han, M.S., D.Y. Jung, C. Morel, S.A. Lakhani, J.K. Kim, R.A. Flavell and R.J. Davis, 2012. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science, 339: 218-222.
- Kojta, I., M. Chacińska and A. Błachnio-Zabielska, 2020. Obesity, bioactive lipids, and adipose tissue inflammation in insulin resistance. Nutrients, 12.
- Yesupatham, A. and R. Saraswathy, 2025. Role of oxidative stress in prediabetes development. Biochem. Biophys. Rep., 43.
- Lingappan, K., 2018. NF-κB in oxidative stress. Curr. Opin. Toxicol., 7: 81-86.
- Yang, Q., A. Vijayakumar and B.B. Kahn, 2018. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol., 19: 654-672.
- Nie, J., Y. Chang, Y. Li, Y. Zhou, J. Qin, Z. Sun and H. Li, 2017. Caffeic acid phenethyl ester (propolis extract) ameliorates insulin resistance by inhibiting JNK and NF-κB inflammatory pathways in diabetic mice and HepG2 cell models. J. Agric. Food Chem., 65: 9041-9053.
- Solinas, G. and B. Becattini, 2016. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab., 6: 174-184.
- Morel, C., C.L. Standen, D.Y. Jung, S. Gray and H. Ong et al., 2010. Requirement of JIP1-mediated c-Jun N-terminal kinase activation for obesity-induced insulin resistance. Mol. Cell. Biol., 30: 4616-4625.
- Huvers, F.C., C. Popa, M.G. Netea, F.H.J. van den Hoogen and C.J. Tack, 2007. Improved insulin sensitivity by anti-TNFα antibody treatment in patients with rheumatic diseases. Ann. Rheumatic Dis., 66: 558-559.
- Hatting, M., C.D.J. Tavares, K. Sharabi, A.K. Rines and P. Puigserver, 2018. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci., 1411: 21-35.
- Donath, M.Y., D.T. Meier and M. Böni-Schnetzler, 2019. Inflammation in the pathophysiology and therapy of cardiometabolic disease. Endocr. Rev., 40: 1080-1091.
- Dimeji, I.Y., K.S. Abass, N.M. Audu and A.S. Ayodeji, 2025. L-Arginine and immune modulation: A pharmacological perspective on inflammation and autoimmune disorders. Eur. J. Pharmacol., 997.
- Chatzianagnostou, K., M. Gaggini, A.S. Florentin, L. Simonini and C. Vassalle, 2024. New molecules in type 2 diabetes: Advancements, challenges and future directions. Int. J. Mol. Sci., 25.
- Bellucci, P.N., M.F.G. Bagnes, G. di Girolamo and C.D. González, 2016. Potential effects of nonsteroidal anti-inflammatory drugs in the prevention and treatment of type 2 diabetes mellitus. J. Pharm. Pract., 30: 549-556.
- Perera, A.P., R. Fernando, T. Shinde, R. Gundamaraju and B. Southam et al., 2018. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep., 8.
- Rohm, T.V., D.T. Meier, J.M. Olefsky and M.Y. Donath, 2022. Inflammation in obesity, diabetes, and related disorders. Immunity, 55: 31-55.
- Wang, M., M. Zhao, J. Yu, Y. Xu and J. Zhang et al., 2022. MCC950, a selective NLRP3 inhibitor, attenuates adverse cardiac remodeling following heart failure through improving the cardiometabolic dysfunction in obese mice. Front. Cardiovasc. Med., 9.
- Tsalamandris, S., A.S. Antonopoulos, E. Oikonomou, G.A. Papamikroulis and G. Vogiatzi et al., 2019. The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol. Rev., 14: 50-59.
- Berbudi, A., S. Khairani and A.I. Tjahjadi, 2025. Interplay between insulin resistance and immune dysregulation in type 2 diabetes mellitus: Implications for therapeutic interventions. ImmunoTargets Ther., 14: 359-382.
- Xu, W., Y. Huang and R. Zhou, 2025. NLRP3 inflammasome in neuroinflammation and central nervous system diseases. Cell. Mol. Immunol., 22: 341-355.
- Seok, J.K., H.C. Kang, Y.Y. Cho, H.S. Lee and J.Y. Lee, 2021. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch. Pharmacal Res., 44: 16-35.
- Yung, J.H.M. and A. Giacca, 2020. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells, 9.
- van der Velden, J.L.J., Y. Ye, J.D. Nolin, S.M. Hoffman and D.G. Chapman et al., 2016. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin. Transl. Med., 5.
- Adeoye, S.W., I.Y. Dimeji, H.J. Lawan, O. Emmanuel, A.T. Samuel, O.D. Oluwakemi and M.B. Zakari, 2024. Synergistic effects of L-arginine and metformin on oxidative stress, inflammation and glucometabolic enzymes in diabetic rats. Trends Appl. Sci. Res., 19: 180-198.
- Herman, R., N.A. Kravos, M. Jensterle, A. Janež and V. Dolžan, 2022. Metformin and insulin resistance: A review of the underlying mechanisms behind changes in GLUT4-mediated glucose transport. Int. J. Mol. Sci., 23.
- Zhao, X., M. Wang, Z. Wen, Z. Lu and L. Cui et al., 2021. GLP-1 receptor agonists: Beyond their pancreatic effects. Front. Endocrinol., 12.
- El-Saadony, M.T., T. Yang, S.A. Korma, M. Sitohy and T.A. Abd El-Mageed et al., 2023. Impacts of turmeric and its principal bioactive curcumin on human health: Pharmaceutical, medicinal, and food applications: A comprehensive review. Front. Nutr., 9.
- Wu, Q., W. Wu, V. Jacevic, T.C.C. Franca, X. Wang and K. Kuca, 2020. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer. J. Enzyme Inhib. Med. Chem., 35: 574-583.
- Zhang, X., Z. Wang, Y. Zheng, Q. Yu and M. Zeng et al., 2023. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases (Review). Int. J. Mol. Med., 51.
- Cui, J., Y. Chen, H.Y. Wang and R.F. Wang, 2014. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccines Immunother., 10: 3270-3285.
- Chen, B., Y. Wang and G. Chen, 2023. New potentiality of bioactive substances: Regulating the NLRP3 inflammasome in autoimmune diseases. Nutrients, 15.
- Zeke, A., M. Misheva, A. Reményi and M.A. Bogoyevitch, 2016. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. Rev., 80: 793-835.
- Alonaizan, R., 2024. Molecular regulation of NLRP3 inflammasome activation during parasitic infection. Biosci. Rep., 44.
- Li, Y., R. Qiang, Z. Cao, Q. Wu, J. Wang and W. Lyu, 2024. NLRP3 inflammasomes: Dual function in infectious diseases. J. Immunol., 213: 407-417.
How to Cite this paper?
APA-7 Style
Dimeji,
I.Y., Jabba,
H.L., Murtala,
N., Ayodeji,
A.S. (2025). Therapeutic Targeting of NLRP3 Inflammasome and JNK Pathways in Inflammation-Induced Insulin Resistance. Asian Science Bulletin, 3(3), 205-223. https://doi.org/10.3923/asb.2025.205.223
ACS Style
Dimeji,
I.Y.; Jabba,
H.L.; Murtala,
N.; Ayodeji,
A.S. Therapeutic Targeting of NLRP3 Inflammasome and JNK Pathways in Inflammation-Induced Insulin Resistance. Asian Sci. Bul 2025, 3, 205-223. https://doi.org/10.3923/asb.2025.205.223
AMA Style
Dimeji
IY, Jabba
HL, Murtala
N, Ayodeji
AS. Therapeutic Targeting of NLRP3 Inflammasome and JNK Pathways in Inflammation-Induced Insulin Resistance. Asian Science Bulletin. 2025; 3(3): 205-223. https://doi.org/10.3923/asb.2025.205.223
Chicago/Turabian Style
Dimeji, Igbayilola, Yusuff, Hamidu Lawan Jabba, Ngabea Murtala, and Adekola Saheed Ayodeji.
2025. "Therapeutic Targeting of NLRP3 Inflammasome and JNK Pathways in Inflammation-Induced Insulin Resistance" Asian Science Bulletin 3, no. 3: 205-223. https://doi.org/10.3923/asb.2025.205.223

This work is licensed under a Creative Commons Attribution 4.0 International License.